
Written by Dr. Judit M. Perez Ortiz Edited by Dr. Marija Cvetanovic
In a tour de force study, a collaborative team of scientists led by Dr. Rudolph Tanzi (Harvard Medical School) and Dr. Huda Zhogbi (Baylor College of Medicine) found a novel relationship between the Spinocerebellar ataxia type 1 gene (ATXN1) and Alzheimer’s disease.
Alzheimer’s disease is the most common neurodegenerative disease and the most common cause of dementia. Its precise etiology remains the subject of intense investigation and debate. Alzheimer’s is a devastating disease. Persons with Alzheimer’s disease experience difficulties thinking and remembering things. As the disease worsens other symptoms begin to appear, such as getting lost easily, not recognizing loved ones, problems with language, and behavioral and psychiatric issues. In the more advanced stages, patients are completely dependent on their caregivers.

Despite extensive research, a specific unifying cause of Alzheimer’s disease has not yet been identified, likely due to its complexity. There are a handful of genes responsible for a rare form of early onset Alzheimer’s disease that affects younger patients. However, the great majority of cases start late in life and have no known underlying cause (termed sporadic). While the major risk to develop Alzheimer’s disease is advanced age, scientists believe that clues to sporadic Alzheimer’s can be found in our genes. In this pursuit, hundreds of “risk genes” have been associated with Alzheimer’s disease and Ataxin-1 (ATXN1) has recently emerged as one of such risk genes. Yet how ATXN1 influence Alzheimer’s disease was not understood.
For decades, ATXN1 had been recognized for its causative role in Spinocerebellar ataxia type 1 (SCA1). Normal ATXN1 gene has in its sequence a repeating string of C-A-G nucleotides (components that make up DNA) one after the other. The cell “reads” these DNA instructions to make ATXN1 protein with a string of glutamines (Q) corresponding to the C-A-G. In the case of SCA1, there is an abnormally long sequence of glutamine repeats (polyQ ATXN1) that leads to disease. The classic symptoms of SCA1 are primarily of coordination of movement, due to damage in an area of the brain called the cerebellum. As the disease worsens, other brain regions become affected and lead to more serious symptoms, such as problems with swallowing and breathing. These are tasks led by a region called the brainstem, which lies between the brain and the spinal cord. While recognized less than the movement difficulties, SCA1 patients also experience difficulties with thinking and memory.
Thinking and memory deficits seen in Alzheimer’s are a result of brain damage in areas that are key to these important processes, the cerebral cortex and hippocampus. Doctors can see this damage using brain imaging (such as MRI) or in the brain tissues in deceased patients. Another disease-defining characteristics of Alzheimer’s brains is the abnormal accumulation of protein aggregates, such as amyloid plaques in these vulnerable brain regions. Because of this, physicians and scientists have long attributed Alzheimer’s to an insult caused by these proteins. Amyloid plaques are composed of a protein called β-amyloid (Aβ, “a-beta”) that is producted In a two-step process. A longer amyloid precursor protein (APP) is “cut” sequentially by β-secretase 1 (BACE1) enzyme, then by γ-secretase enzyme. Brains of Alzheimer’s patients have higher levels of BACE1 enzyme than healthy brains. This observation begs the question, what regulates BACE1 levels in the brain? And why does this occur in the specific brain regions that are affected by Alzheimer’s?
A new study led by Dr. Rudolph Tanzi (Harvard) and Dr. Huda Zhogbi (Baylor) uncovered a novel mechanism on how Aβ production is regulated by ATXN1 gene. First, the scientists asked whether patients with Alzheimer’s share the same type of mutation in the ATXN1 gene as that seen in patients with SCA1. They found no difference between the C-A-G repeat region in the Alzheimer’s group compared to healthy controls, leading to the conclusion that the genetic mutation in ATXN1 that causes SCA1 is not linked to Alzheimer’s. How is the ATXN1 gene linked to Alzheimer’s, then?
In the journey to studying SCA1, researchers developed mice that lack the equivalent of human ATXN1 gene (the mouse gene is spelled Atxn1). Mice lacking Atxn1 do not develop the motor issues as seen in SCA1, but do demonstrate hippocampal-dependent learning and memory deficits. Inspired by the parallel between this observation and the hippocampal deficits seen in Alzheimer’s, the group then asked whether nonfunctional or missing Atxn1 in the brain would lead the way to an answer.
To study this, the scientists examined how lack of Atxn1 alters disease pathology in a mouse model of Alzheimer’s disease (APP/PS1, or “AD mice” for this article). The “AD mice” express in their brains two genetic mutations associated with Alzheimer’s disease in humans that increase production of Aβ and amyloid plaque formation. What they found was astounding. The brains of AD mice lacking Atxn1 showed worse Alzheimer’s related pathology than brains of AD mice with normal levels of atxn1!
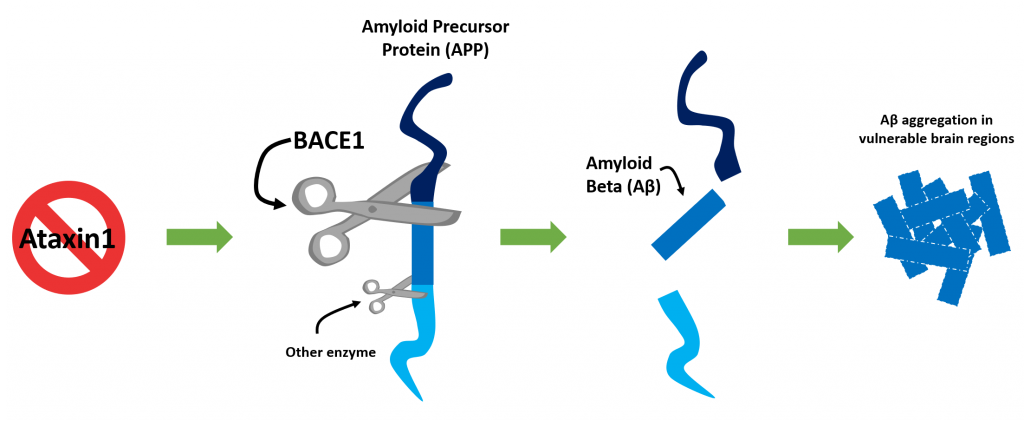
To better understand why this is the case, the investigators looked into the components of the machinery that processes APP into amyloid peptides. They found that BACE1, the enzyme that cleaves APP to make Aβ, is more abundant in the brains of mice lacking Atxn1 than those with normal levels of Atxn1. It turns out that without ATXN1 protein around, other proteins it works with to orchestrate expression of genes is disturbed. Consequently, there is an inappropriate increased production of BACE1. This is a previously undescribed process on how BACE1 abundance is regulated and an important one for informing potential therapeutic strategies for Alzheimer’s.
A critical finding is that BACE1 increase was unique to the brain areas affected in Alzheimer’s disease, but not in the brain regions affected in SCA1. In other words, one gene (Atxn1) plays different roles in different regions of the brain, and poses a differential risk to two distinct neurodegenerative diseases. This insight is a major contribution to the fields of genetics and neurodegeneration. Adding to the effects genetics has on disease risk, the investigators found that the changes observed were unique to the adult (mature) brain, and absent in the developing (immature) brains. While the Atxn1 gene doesn’t change in the lifetime of the individual, the timing of effects it has on the brain is age-dependent.
Central to memory formation in adult brains is the generation of new neuronal cells in the hippocampus (called hippocampal neurogenesis), which is affected in Alzheimer’s. Previously, another group led by Dr. Marija Cvetanovic (University of Minnesota) demonstrated that either lack of Atxn1 or the presence polyQ ATXN1 mutant can impair hippocampal neurogenesis. The present work added to the story by showing that this deficit is likely mediated by increased levels of BACE1 in either scenario. What was surprising is that BACE1 abundance was not increased by the same mechanism. Intriguingly, while Atxn1 loss did not affect BACE1 levels in the cerebellum, the SCA1-related polyQ ATXN1 did. Further, while absence of ATXN1 or presence of polyQ ATXN1 affected hippocampal neurogenesis, only the latter also cause neuronal death in a region of the hippocampus. A critical revelation became evident in this study – it is not only the gene, but the type of genetic abnormality that determines the manner in which it affects the brain. While lack of Atxn1 causes the above mentioned changes, the C-A-G repeat mutation seen in SCA1 imparts new abnormal functions.
The significance of this work is twofold and addresses enigmatic questions in the field of neurodegeneration. First, a single gene (ATXN1) can play different roles. If ATXN1 hosts a C-A-G mutation, this leads to SCA1 disease. If ATXN1 is deficient, this increases Alzheimer’s related pathology. Second, mutant ATXN1 uniquely affects SCA1 vulnerable brain regions, while absence of ATXN1 spares those regions. Instead, lack of ATXN1 increases susceptibility to Alzheimer’s disease. It seems that normal ATXN1 provides protection for Alzheimer’s disease.
Key Terms
Amyloid: A type of protein that tends to aggregate and “stick” together.
Brainstem: A complex region of the nervous system that seats between the brain and the spinal cord. Among other functions, neurons that help speech, swallowing, regulating facial movements, reside here. Insults to the brainstem result in any of these being affected.
Gene: A sequence of “instructions” in the DNA that is used to make RNA or proteins that carry out functions in the cell.
Gene Expression: when the information in a DNA sequence (a gene) is “read” to make its corresponding protein.
Hippocampus: a region of the brain important for emotion, learning, and memory.
polyQ ATXN1: The mutant (abnormal) form of ATXN1. polyQ ATXN1 causes SCA1 disease in humans.
Conflict of Interest Statement
The authors and editor declare no conflict of interest.
Citation of Article Reviewed
Suh J, Romano DM, Nitschke L, Herrick SP, DiMarzio BA, Dzhala V, Bae JS, Oram MK, Zheng Y, Hooli B, Mullin K, Gennarino VA, Wasco W, Schmahmann JD, Albers MW, Zoghbi HY, Tanzi RE. Loss of Ataxin-1 Potentiates Alzheimer’s Pathogenesis by Elevating Cerebral BACE1 Transcription. Cell. 2019 Aug 22;178(5):1159-1175.e17. https://www.ncbi.nlm.nih.gov/pubmed/31442405.

