
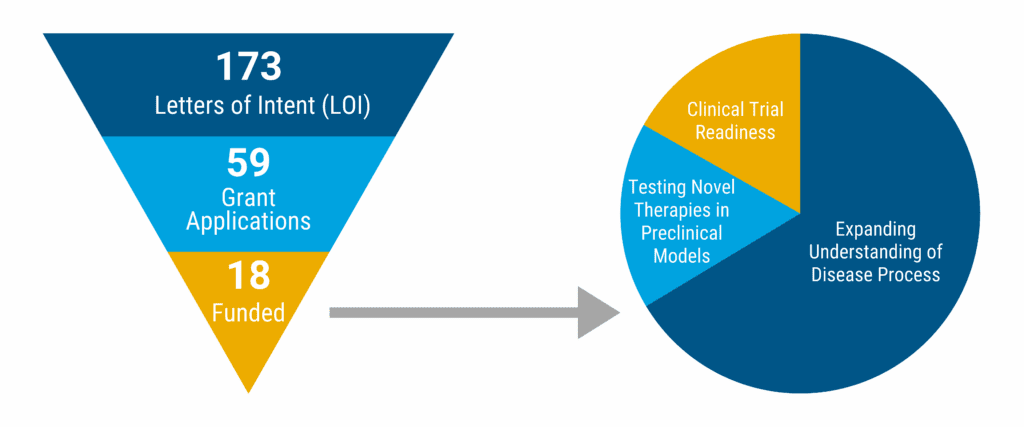
NAF Awarded a Record Number of Projects in 2024
18 Studies Funded Totaling $845k Funded!
We had our most competitive year ever for Ataxia research grant applications! As a result of the generosity of our donors, NAF was able to fund a record number of Ataxia research grants in 2024!
To receive an Ataxia research grant from NAF, researchers go through a 4-step process:
- Submit a Letter of Intent (LOI) – This is a brief overview that gives information about the goal of their research study.
- LOIs are Reviewed – A panel of Ataxia experts review and score each LOI for scientific merit. They determine which LOIs will be invited to submit a grant application.
- Submit Grant Application – Studies which received a high enough score to move to the next step of the process are invited to submit a full application. The grant application requires more time and details about their project.
- Grants Applications Reviewed and Recommended for Funding – Each application receives 3 separate scientific reviews. NAF has 75 scientific reviewers who evaluate and score each full application that is received. Top-scoring grant applications are recommended for funding.
We received 173 Letters of Intent, which was a 72% increase in submissions versus 2023! We’re happy to know that more Ataxia researchers are seeking funding for their studies! Thank you to all the researchers who submitted an LOI or application as well! Keep up the great work that is SO important to the Ataxia community!
59 researchers were invited to submit a grant application. We’re proud to announce that we funded a record number of research grants – 18 in total! A full list of those studies is included below.
A huge thank you to our scientific reviewers who volunteer their time and expertise to make sure that the best science to accelerate Ataxia treatment development is funded!
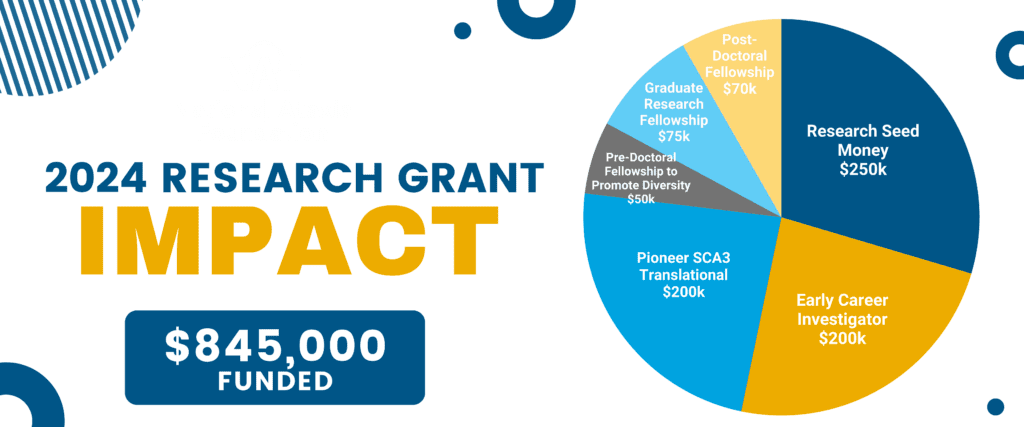
For 2024, NAF funded the following research:
- 5 Research Seed Money Grants – totaling $250,000
- 4 Early Career Investigator Awards – totaling $200,000
- 3 Graduate Research Fellowships – totaling $75,000
- 2 Pioneer SCA Translational Award – totaling $200,000
- 2 Post-Doctoral Fellowship – totaling $70,000
- 2 Pre-Doctoral Fellowship to Promote Diversity in Ataxia Research – totaling $50,000
To learn more about each type of research study, visit www.ataxia.org/researcher-resources.
A full list of the research studies and their lay summaries are available below.
Lay Summaries
Research Seed Money Grants
$50,000 – 1-Year Grant
88 LOIs Received | 22 Full Applications Reviewed | 5 Funded

Dr. Nicolas Charlet
Institut de Genetique et de Biologie Moleculaire et Cellulaire
Research Title: A common RNA toxicity mechanism causes neuronal cell dysfunctions in SCA10, 31 & 37
Ataxia Type: SCA10, SCA31 and SCA37
Spinocerebellar ataxia 10, 31 and 37 are caused by mutations of strikingly similar AT-rich sequences. These mutations are suspected to be pathogenic at the RNA level, however, through an ill-defined mechanism.
Importantly, we found that these AU-rich RNA repeats bind and capture specific RNA binding proteins, essential for neuronal cell physiology. Thus, we propose to explore whether detention of these RNA binding proteins would result in their lesser availability, resulting in specific mRNA metabolism changes leading to neuronal cells dysfunctions and death.
Overall, this National Ataxia Foundation Seed funding will help to develop relevant cell and animal models and clarify the pathogenic mechanisms at play in SCA10, 31 and 37, an essential first step to identify relevant biomarkers and therapeutic approaches for these neurodegenerative diseases.

Dr. Beatriz Ibarra-Molero
Universidad de Granada
Research Title: Towards the development of precision treatments for different types of ataxia linked to transglutaminase 6
Ataxia Type: SCA35, Gluten Ataxia
Our research focuses on Transglutaminase-6 (TG6) a protein that plays a key role in, at least, two forms of Ataxias: Spinocerebellar Ataxia type 35 (SCA35), a rare inherited ataxia caused by specific alterations in TG6-encoding gene, and gluten ataxia (GA) which is suffered by patients genetically susceptible to gluten and involves a pathological immune response producing antibodies against TG6.
With the NAF support, we aim to perform a comprehensive characterization of TG6-pathogenic variants and TG6 interactome. Our results will not only shed light on the molecular mechanisms linking TG6 dysfunction with ataxias but will also pave the way to design novel therapeutical avenues to treat and prevent the devastating neurological manifestations caused by these diseases.

Dr. Jordi Magrane
Cornell University
Research Title: Selective frataxin deficiency in glial cells in vivo in novel mouse models of Friedreich’s ataxia
Ataxia Type: Friedreich’s Ataxia
Contribution of glial cells to Friedreich’s ataxia (FRDA) pathophysiology remains mostly unexplored. We propose to investigate the involvement of four types of glia cells to the neuropathy present in FRDA using two new mouse models. We will specifically deplete frataxin from oligodendrocytes and astrocytes (in the central nervous system), and Schwann cells and satellite cells (in the peripheral nervous system) and investigate its impact on the early mouse nervous system development and neurobehavior. Moreover, we will assess both the consequences of frataxin deficiency within glia cells (cell autonomous component) and a potential non-cell autonomous effect over neurons in vivo. Successful completion of this project will address the contribution of glial cells to the nervous system pathology in FRDA, which is critical for the development of therapies to prevent or reverse white matter damage and gliosis and likely to influence neuronal survival.

Dr. James Orengo
Baylor College of Medicine
Research Title: Studying motor neuron disease in Spinocerebellar Ataxia, type 1 with novel zebrafish models
Ataxia Type: SCA1
Spinocerebellar Ataxia type 1 (SCA1) is a devastating neurodegenerative disease, leading to premature death typically by the third to sixth decade of life. While the hallmark of SCA1 is loss of motor coordination or ataxia, at the end stage of disease premature death results from complications secondary to muscle weakness, including breathing and swallowing difficulties. Substantial effort has been invested in determining the mechanisms leading to cerebellar degeneration in SCA1; however, degeneration of the cerebellum cannot account for the muscle weakness, and breathing and swallowing difficulties, which ultimately causes premature death. These symptoms are time locked with the degeneration of another type of brain cell called the motor neuron. The goal of this proposal are to use newly developed zebrafish models of SCA1, which allow for the production, of the toxic protein made in SCA1, to only occur in motor neurons and to determine whether this leads to motor neuron degeneration, muscle wasting and premature death in zebrafish.

Dr. Pedro Rodriguez Cruz
National Center for Genomic Analysis (CNAG)
Research Title: Hereditary Ataxia in underrepresented populations of West Africa: increasing diversity, equity and inclusion in ataxia research
Ataxia Type: Hereditary Ataxias
Studies in different populations have shown that the genetic causes of hereditary ataxias are highly heterogenous across the world. However, there exist a significant knowledge gap in Sub-Saharan Africa where populations of African genetic ancestry remain genetically unexplored. Factors such as migration, population genetics and founder effects (an individual spreading a faulty gene in successive generations) strongly influenced the landscape of hereditary ataxia, with some specific ataxia subtypes being common in one area but extremely rare in others. With Africa’s population set to double by 2050, it is crucial to address this knowledge gap and explore the unique genetic, ethnic, and geographical factors influencing hereditary ataxia in populations with African genetic ancestry. Our project aims improve our understanding of the hereditary ataxias in Senegal and West Africa, discover new causes of ataxia, and seed collaborations and interest in the field by developing a regional ataxia network.
Early Career Investigator Awards
$50,000 – 1-Year Grant
32 LOIs Received | 11 Full Applications Reviewed | 4 Funded

Dr. Adam Avery
Oakland University
Research Title: A cerebellum-specific ß-III-spectrin protein interaction network for SCA5 insights
SCA5
Ataxia Type: SCA5
Mutations in the SPTBN2 gene encoding the protein β-III-spectrin cause SCA5. It is known that β-III-spectrin is required for the health of Purkinje neurons in the cerebellum. However, the protein-protein interactions required for β-III-spectrin to support Purkinje neuron health are not well defined, limiting our understanding of SCA5 disease. In this project, using cerebellum from healthy mice, or a SCA5 mouse model, we will perform an unbiased protein-protein interaction screen to 1) identify novel β-III-spectrin protein interactors, and to 2) gain insight into how SCA5 mutations impact β-III-spectrin protein complexes.

Dr. Allison Hilger
University of Colorado-Boulder
Research Title: Respiratory Incoordination as a Driving Mechanism in Ataxic Dysarthria
Ataxia Type: SCAs, FA
Damage to the cerebellum in spinocerebellar ataxia impairs the coordination of movement across the body, including the rapid and precise movements needed for fluent and intelligible speech. In this study, we are measuring coordination between breathing and voicing, referred to as respiratory-laryngeal coordination. Breathing is the driving force for speech, and improvement in breath control could have high potential for improving speech in Ataxia.

Dr. Louisa Selvadurai
Monash University
Research Title: A scale to evaluate the neuropsychiatry of cerebellar diseases
Ataxia Type: SCAs, ARCA, ILOCA, MSA-C, CANVAS
People with cerebellar diseases may experience difficulties with their emotions and behaviour as a result of changes to the cerebellum. These difficulties, referred to as neuropsychiatric symptoms, are at risk of being overlooked in the clinic but may lead to problems with wellbeing, relationships, employment, and engagement with healthcare. There is therefore an urgent need to find a better way to identify, measure, and manage neuropsychiatric symptoms in this population.
The Cerebellar Neuropsychiatric Rating Scale (CNRS) was developed to meet this need by the Schmahmann Lab at Massachusetts General Hospital. In this project, Massachusetts General Hospital and Monash University will collaborate to develop and test a final version of the CNRS, so that it can be used in the clinic and in research studies and ultimately provide improved care and better outcomes for affected individuals and their families.

Dr. Andreia Teixeira-Castro
University of Minho
Research Title: Decoding the serotonergic circuitry in the Machado-Joseph disease mouse brain
Ataxia Type: SCA3
Previously, we found that citalopram, an antidepressant commonly used for treatment of depression that increases the neurotransmitter serotonin in the brain, restores motor function, and decreases mutant ATXN3 aggregation, leading us to believe it is a safe therapeutic compound to be used for SCA3/MJD. In this project, we aim at studying the communication between the brain cells that produce serotonin in the brain (Raphe nuclei neurons) with the cells of the cerebellum (the deep cerebellar nuclei neurons), highly affected in MJD. We will see if the activation of the serotonergic neurons of the Raphe in the brain of MJD mice can restore their motor function, decrease aggregation of the mutant protein, and attenuate neuronal demise. We believe that the outcomes of this proposal will lead to new treatments for MJD and perhaps other Ataxias.
Graduate Research Fellowship
$25,000 – 1-Year Grant
34 LOIs Received | 14 Full Applications Reviewed | 3 Funded

Mr. Yin Chun Cheng, Vincent
University of Oxford
Research Title: Elucidating developmental gene expression changes in Spinocerebellar Ataxia 44
Ataxia Type: SCA44
Spinocerebellar Ataxia (SCA) is a group of genetic disorders that impair the cerebellum, the part of the brain responsible for movement and balance, leading to a progressive decline in motor functions. Although it is believed that SCA is due to the cerebellum deteriorating over time, recent research suggests that the problem may originate from disrupted cerebellar development early in life. This project aims to explore the mechanisms that cause abnormal development of the nerve cells in the cerebellum and thereby contribute to the progression of SCA. Ultimately, we hope that the knowledge gained through thus study will help to identify new therapeutic strategies for SCA patients.

Ms. Alexa Putka
University of Michigan
Research Title: Elucidating Cholesterol Impairments in Spinocerebellar Ataxia Type 3: A Mechanism Underlying Oligodendrocyte Maturation Deficits
Ataxia Type: SCA3
Our research investigates oligodendrocytes, brain cells that wrap neurons with insulation called myelin to speed up electrical transmission in the brain. Our lab recently found that oligodendrocytes have trouble maturing from their earliest state to their mature myelinating state in Spinocerebellar Ataxia type 3 (SCA3). We know cholesterol is an essential component of myelin and is required for oligodendrocyte maturation. In this proposal, we will explore the cholesterol pathway in SCA3 patient postmortem tissue and mouse models to determine the relationship between cholesterol and reduced oligodendrocyte maturation. This work will help us better understand the changes underlying SCA3 disease so that we may better intervene.

Mr. Tyler Thaxton
University of Chicago
Research Title: The functional domains of a1ACT and their impact on spinocerebellar ataxia type 6
Ataxia Type: SCA6
Our lab discovered that a protein expressed in an embedded reading frame inside CACNA1A, termed a1ACT, contains a repeating tract of the glutamine amino acid, called a polyglutamine tract. Spinocerebellar Ataxia type 6 (SCA6) is caused by a mutation in the CACNA1A gene, wherein the polyglutamine tract is expanded past a normal range, which may affect any resulting protein’s ability to function properly. Our mouse models with reduced a1ACT expression exhibit ataxic-like symptoms. However, we do not fully understand how a1ACT functions, only that the polyglutamine tract expansion causes negative downstream effects. I am seeking to elucidate what domains a1ACT uses to better understand its disease-causing, polyglutamine expanded counterpart. Any knowledge we can gain on how a1ACT performs in either its normal or disease state will help us understand how SCA6 occurs and provide more clues to novel therapeutic options.
Pioneer SCA3 Translational Research Award
$100,000 – 1-Year Grant
9 LOIs Received | 6 Full Applications Reviewed | 2 Funded

Dr. Maria do Carmo Costa
University of Michigan
Research Title: Investigating the efficacy of aripiprazole related compounds as a therapeutic option for SCA3
Ataxia Type: SCA3
An attractive route to preventive therapy for SCA3 is to reduce levels of the toxic ATXN3 protein. We previously identified the atypical antipsychotic aripiprazole as being effective to reduce the abundance of mutant ATXN3 protein in multiple cellular and ex vivo models of SCA3, as well as in vivo in SCA3 flies and in brains of SCA3 transgenic mice. However, aripiprazole shows pleiotropic effects and is not potent enough to improve motor dysfunction in SCA3 mice. In this study, we will identify aripiprazole related compounds as potential therapeutic agents for SCA3 by developing novel potent compounds that reduce levels of pathogenic ATXN3 in human neurons, and by evaluating the in vivo therapeutic efficacy of the novel compounds to mitigate pathogenic ATXN3 toxicity in SCA3 transgenic mice.

Dr. Jennifer Faber
German Center for Neurodegenerative Diseases (DZNE)
Research Title: Biomarker enriched data-driven disease model of SCA3
Ataxia Type: SCA3
In Spinocerebellar Ataxia type 3 (SCA3), fluid and imaging biomarkers change already before the clinical onset of the ataxia. A data-driven disease model enriched with such biomarker information is a prerequisite to better understand the temporal dynamics of changes in the entire disease course including the so called “Biomarker stage” before clinical manifestation. It will allow the identification of progression as well as stratification markers and eventually, personalized predictions. The availability of such a data-driven integrated staging model of SCA3 will directly inform planning and design of clinical trials.
Post-Doctoral Fellowship
$35,000 – 1-Year Grant
11 LOIs Received | 6 Full Applications Reviewed | 2 Funded

Dr. Ilaria Balbo
Columbia University
Research Title: Testing cerebellar circuit restoration therapies for Spinocerebellar Ataxia 1 (SCA1)
Ataxia Type: SCA1
Individuals with spinocerebellar ataxia 1 (SCA1) suffer from debilitating loss of movement control. Yet, no existing therapies are available, partially due to an incomplete understanding of the brain circuit alterations responsible for these symptoms. Prior research in animal models has elucidated that, before degenerating in the late stage of the pathology, Purkinje cells (PCs) initially prune their excitatory synapses, including climbing fiber (CF)-PC synapses. Therefore, targeting these circuit changes in a stage-specific manner holds promise for restoring circuit function in SCA1 and designing future clinical trials. I aim to test the hypothesis that promoting PC-CF synaptic formation with gene therapy and/or restoring cerebellar rhythm with a wireless closed-loop stimulation differentially improves ataxia symptoms in the early vs. late stages of the pathology in a SCA1 mouse model.

Dr. Emmanuel Labrada
University of Minnesota
Research Title: SCA1 progressively impacts on the function of the motor network striatum- cerebellum
Ataxia Type: SCA1
Spinocerebellar Ataxia type 1 (SCA1) is a neurodegenerative disease that results from the expansion of CAG repeats in the ATAXIN-1 gene. This disease primarily targets the cerebellum, causing severe degeneration of Purkinje cells (PCs) and cerebellar nuclei neurons. Recent studies suggest that SCA1 extends its impact beyond the cerebellum, affecting neuronal populations in the striatum and cortex. Our research aims to explore the functional changes within the motor network of SCA1 to enhance our understanding of network function in healthy brains and its dysfunction in SCA1, ultimately inspiring therapeutic approaches.
Pre-Doctoral Fellowship to Promote Diversity in Research
$50,000 – 2-Year Grant
2 Full Applications Reviewed | 2 Funded

Mr. Juan Mato
University of Michigan
Research Title: Characterizing peripheral neuropathy onset and progression in SCA3 mouse models
Ataxia Type: SCA3
Spinocerebellar Ataxia type 3 (SCA3) is the most common dominantly inherited ataxia and is most often associated with symptoms like uncoordinated movement, difficulty swallowing, and gait abnormalities. However, more than half of SCA3 patients experience peripheral neuropathy symptoms of reduced sensation or burning pains in distal limbs that take a heavy toll on quality of life. My research will interrogate onset, progression, and pathogenic mechanism of peripheral neuropathy in mouse models of SCA3 in order to pave the way for future therapeutic development for peripheral SCA3 symptoms.

Ms. Sabrina Phanor
Columbia University
Research Title: PUMILIO2 deficiency: understanding a new ataxia gene and its role in cerebellar dysfunction in mice
Ataxia Type: PUM1/2 Associated Ataxias (SCA47)
Several years ago, we found that mutations in the RNA-binding protein, PUM1, cause at least two forms of SCA47: a severe, early-onset neurodevelopmental syndrome, and a very mild, late-onset pure cerebellar ataxia. As we studied this disease in patient cell lines and mouse models, we noticed that the levels of the PUM1 homolog, PUM2, are affected by the loss of PUM1 function, raising the possibility that PUM2 mutations might also cause ataxia. Therefore, we sought and found fourteen patients with different mutations in PUM2 who present with various phenotypes that parallel to SCA47, ranging from neurodevelopmental problems to young adult-onset cerebellar ataxia. This fellowship will be used to investigate PUM2 mutant mice to understand this new SCA.
View Past Years Funded Research
Thank You Donors!
The generosity of our donors allows NAF to focus on our mission to accelerate the development of treatments and a cure while working to improve the lives of those living with Ataxia. We could not offer this service to the Ataxia research community without you!

