
Research is rapidly moving from the bench to the bedside to treat neurological inherited disorders of all types, including spinocerebellar ataxias. SCAsource has previously gone over the science behind ASO therapy. These diseases share a common theory that the DNA mutation leads to the formation of an altered protein that is toxic. ASO therapy is meant to stop the formation of the toxic protein by “shooting the messenger”.
What is involved in these clinical trials?
To see what might happen in ataxia trials, let’s look at ASO trials happening right now in related polyglutamine diseases. In Huntington’s disease (HD), there are two programs that are currently in clinical trials. Regulatory authorities view ASOs as drugs and require that the product be shown to be both safe and effective in patients.
ASOs cannot be given as pills and they are currently injected into the spinal fluid. This is called intrathecal administration to get the drug directly in the fluid space where it can circulate back to the brain. Patients in phase 1 studies in HD are asked to have up to 7 injections and one phase 3 program requires injections every second month for 2 years. This involves a large commitment to the study and is asking a lot from patients and their families.
The only published phase 1 double-blind, placebo-controlled study in HD (Tabrizi et al., New England Journal of Medicine, 2019) has identified that a series of 4 injections were safe. They measured changes of the “bad” protein in the spinal fluid as a proof of concept that ASOs could lower protein levels. The good news was that they found that there was a dose-related reduction in this protein of about 40%. Patients from this study were offered “open label” monthly injections and this has shown a 60% reduction in the abnormal protein according to a recent presentation. Open label extensions are when patients can continue taking a drug after the formal time of the clinical trial is over.

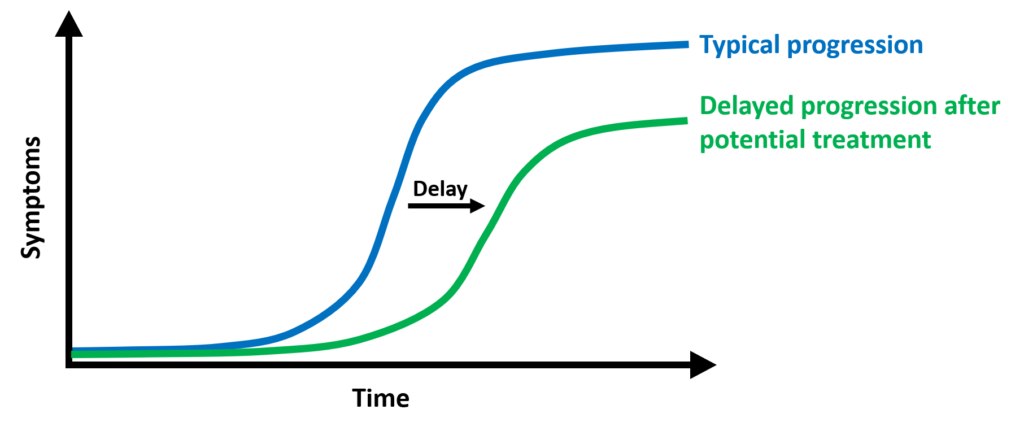
So, what does success mean?
The phase 3 studies that are currently ongoing in HD are designed to see if there is a slowing of disease progression. This is being measured by assessing motor, cognitive and behavioral symptom change over time. Changes occur slowly in HD and SCA. Therefore, large numbers of patients are required over a relatively long study time.
The bottom line is that a successful study that shows slowing disease progression is likely to mean that the patients may not experience any obvious improvement while receiving the treatment and that they will continue to have progressive symptoms over time. Hopefully, this will be at a slower rate compared to the placebo group. Since there are no treatments available for SCA or HD, this will be welcome. It is by no means considered to be a cure or likely to stop the progression. True cures in medicine are rare, where a cure is defined as a drug ending disease.
In the HD research community, we are asking questions that include:
- Is it a good idea to reduce the good protein that is part of our normal brain chemistry? In the current phase 3 study, the ASO reduces both the “good” and the “bad” HD protein. Another program in phase 1 uses an ASO that only reduces the “bad” protein.
- When is the best time to use ASO therapy? Since these conditions are associated with nerve cell damage and loss, it makes sense to use these types of therapy very early, even before damage occurs. This will mean that patients with moderate or advanced symptoms may not be good candidates for ASO therapy.
- Should we consider treatment in people who have had predictive genetic testing before symptoms start? This is being actively discussed but it is too early to consider this. We have to show that ASOs are safe and effective in symptomatic patients. We need to have good measures to determine if treatments are working. Regulatory authorities have required evidence that treatments have a positive effect on patients lives. This may be difficult to show in a short study. We must consider that it takes patients decades to get these diseases: slowing or stopping this could take just as long.
We can only figure out the answers to these questions in clinical trials. The goals of these trials are to improve people’s quality of life. To do this we need information from real people with these diseases, and not just models of disease. This is a process that will take time but will tell us which approach has the most promise and is worth pursuing faster. Thus, the patients and families at this point are just as important as the researchers in lab coats working together to treat these diseases.
If you would like to learn more about clinical trials, take a look at this resource by the FDA or our previous Snapshot on the subject.
Snapshot written by Dr. Mark Guttman and edited by Dr. Ray Truant.

